
Can Biology Survive Data Compression
A taxonomy of preclinical biology’s information loss, and why it is not correctable downstream

Biology's central problem is not data. It is inference: moving from what can be measured to what actually causes disease. Hamiltonian Biology examines that gap — why it exists, how deep it runs, and what closing it would require.
____
Most translational failures are diagnosed as failures of biology: the mouse did not predict the human, the model did not generalize, the target was not as important as we thought. This diagnosis is almost always incomplete. The deeper failure is one of measurement, and measurement failures have a specific character that biological complexity alone does not explain.
In information theory, the data processing inequality states a simple and irreversible fact: if a signal passes through a processing step, no downstream operation can recover information that was discarded upstream. Applied to the preclinical measurement pipeline, this is not a theoretical concern. It is the structural description of why drug after drug clears animal studies and fails in patients. The information that would have predicted failure was present in the biology. It did not survive the compression.
But the data processing inequality, on its own, is not the interesting claim. Information loss is inevitable in any measurement system. The interesting claim is about the character of the loss: specifically, that the preclinical measurement stack was not designed to minimize clinically relevant information loss. It was designed to minimize experimental noise and maximize regulatory legibility, and those two optimization targets are nearly orthogonal to clinical predictive content. The compression was designed for the wrong distortion metric.
Rate-distortion theory formalizes this precisely. For any lossy compression scheme, the optimal representation is defined by the distortion function: the penalty you assign to different kinds of errors. Compress a signal while penalizing experimental variance, and you get tumor volume curves and survival endpoints. Compress the same signal while penalizing loss of mutual information with patient treatment response, and you get something the current preclinical stack does not resemble. Both are valid compression schemes. Only one is useful for the task at hand.
What follows is a taxonomy of what gets discarded, organized not by biological scale but by causal distance from clinical outcome. The goal is not to catalogue the inadequacy of animal models, which is well-trodden ground, but to be precise about the structure of the information loss — because precision about the structure is the only path to understanding what a different measurement design would need to preserve.
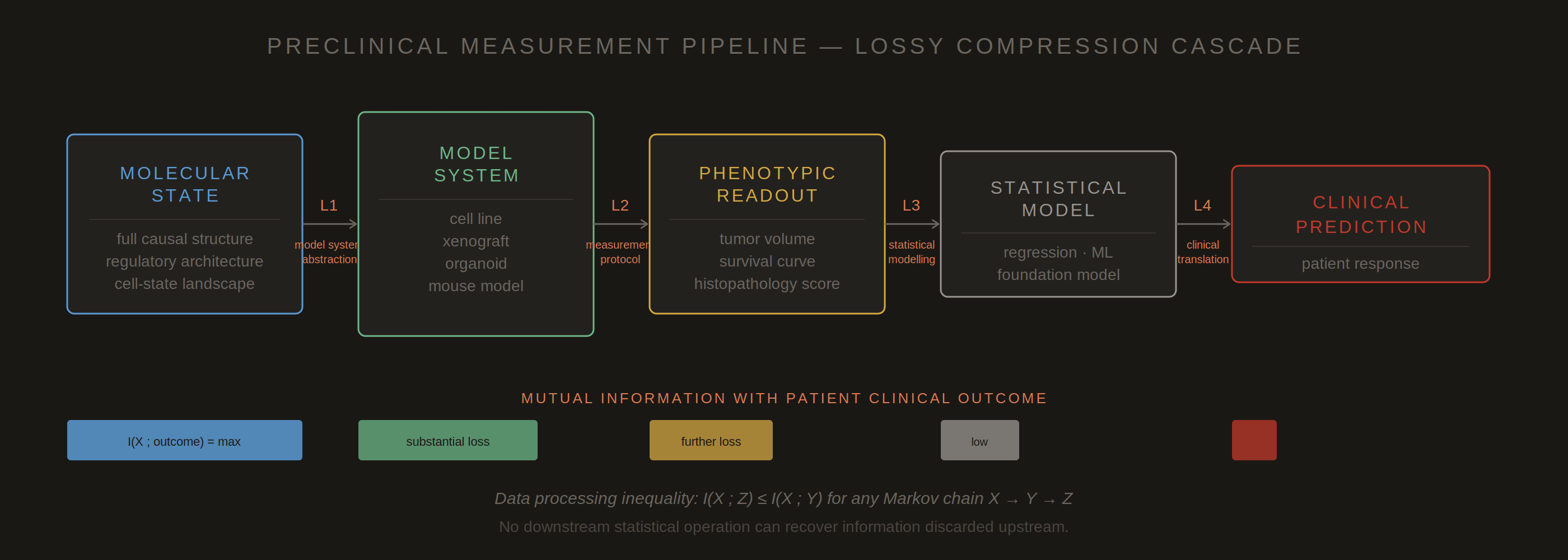
Figure 1. The preclinical measurement pipeline as a cascade of lossy compression steps (L1–L4). At each stage, information present in the upstream biological state is irreversibly discarded. The mutual information between the molecular state and the clinical outcome decreases monotonically with each compression step. The data processing inequality guarantees that no downstream statistical operation — however sophisticated — can recover what was lost upstream. The critical question is not whether information is lost, which is unavoidable, but whether the compression was designed to preserve clinically relevant signal. Standard preclinical protocols were optimized for reproducibility and regulatory legibility, not for this.
I. What Gets Discarded at the Cellular Level
The single-cell heterogeneity problem
A tumor is not a uniform population. It is a community of cells occupying distinct transcriptional states: some proliferating rapidly, some quiescent, some already engaged in the transcriptional programs that will produce resistance to whatever intervention arrives next. Bulk measurements, whether phenotypic or even bulk transcriptomic, compress across this distribution. What survives is the mean. What gets discarded is the tail.
The clinical consequence of compressing out the tail is not subtle. In most therapeutic contexts, the clinical outcome is determined not by the average response but by the behavior of the least sensitive subpopulation. A drug that eliminates ninety-five percent of a tumor’s cells while leaving five percent intact has not achieved ninety-five percent of a cure. It has selected for a resistant clone. The subsequent disease trajectory is governed by the biology of that five percent, which was invisible in the bulk readout that declared success.
The compression filter that the field applied was one that systematically discarded exactly those features most likely to determine clinical outcome.
The EGFR inhibitor story in non-small cell lung cancer is the canonical illustration. Early response rates to first-generation EGFR inhibitors were striking, and bulk tumor measurements reflected genuine cytoreduction. What the bulk measurements could not resolve was the T790M resistance mutation, already present at low frequency in most patients at baseline, invisible against the background of the sensitive majority population. The drug compressed the sensitive population toward detection limits and amplified the resistant minority. The readout that had declared efficacy had been averaging over a distribution that contained its own refutation.
This is not a failure of sequencing depth or statistical power. It is a failure of measurement design. The readout was optimized to detect population-level response, which it did correctly. The clinically relevant variable was subpopulation-level resistance capacity, which the readout could not encode by construction.
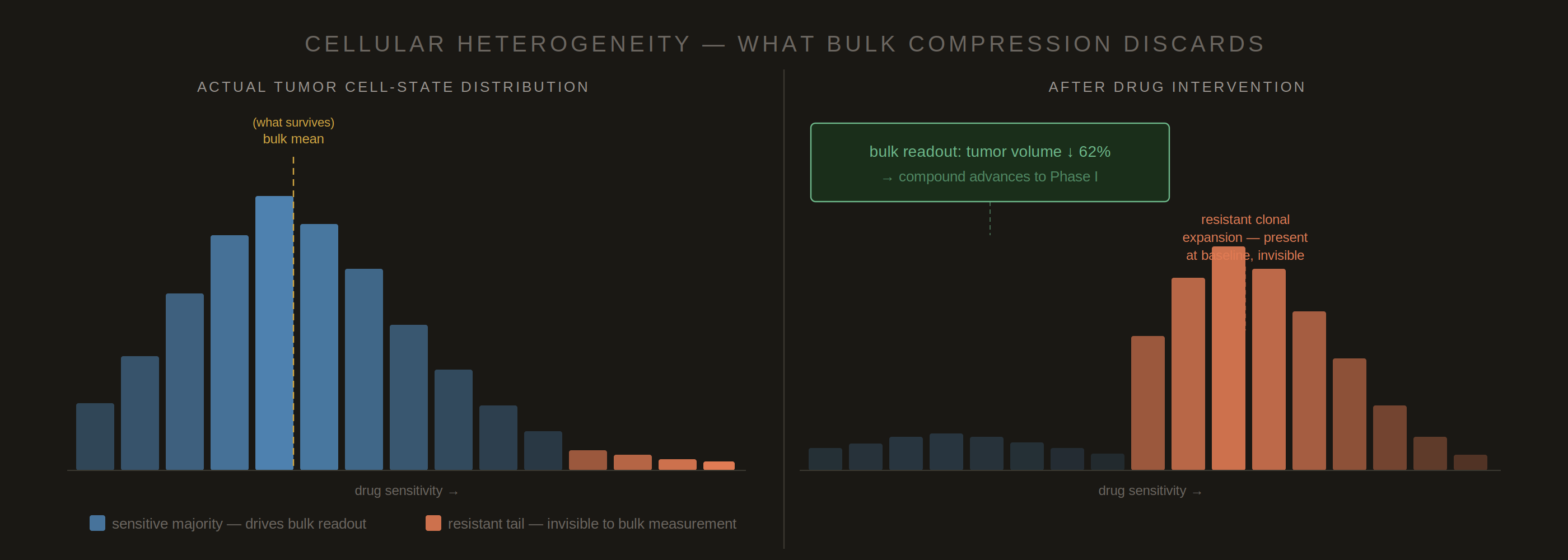
Figure 2. Bulk measurement compresses a heterogeneous cell-state distribution into a single summary statistic. After drug treatment, the sensitive majority is depleted and the resistant tail expands — but the bulk readout records only mean cytoreduction and declares success. The clinically relevant variable is the resistant subpopulation’s dynamics, which were never encoded in the readout. The T790M resistance mutation in EGFR-inhibited non-small cell lung cancer is the canonical case: present at low frequency at baseline, invisible in bulk measurement until relapse. No downstream analysis can recover what was never measured.
II. What Gets Discarded at the Microenvironmental Level
The absent immune compartment
Standard xenograft models use immunocompromised mice. This is not an experimental limitation that researchers have overlooked. It is a design requirement: human cells engrafted into an immunocompetent mouse would be rejected. The practical consequence is that the entire immune compartment — T cell architecture, myeloid polarization states, cytokine communication networks, and the physical organization of immune-tumor interactions — is either absent or profoundly distorted in the model system that is supposed to predict human response.
For a large class of drugs, this is not a nuisance variable to be controlled for. It is the primary mechanism of action. Checkpoint inhibitors do not kill tumor cells directly. They release T cell suppression so that the endogenous immune system can do what it was already trying to do. Measuring checkpoint inhibitor efficacy in a model with no functional T cell compartment is not measuring a weaker version of the clinical signal. It is measuring a different biological process that happens to share a molecular target.
The early checkpoint inhibitor failures in standard mouse models illustrate this precisely. PD-1 blockade appeared unreliable and context-dependent in xenograft systems because the mechanism of action required the very compartment the model had eliminated. The compression step that made the model tractable — immunocompromising the host — also discarded the information that would have predicted clinical efficacy. What survived into the readout was a residual cytotoxic signal from immune-independent effects, which was real but was not the phenomenon of interest.
The stromal architecture presents an analogous problem. Tumors in patients are embedded in a mechanical and biochemical microenvironment that regulates drug penetration, oxygen availability, metabolic substrate access, and the physical constraints on cell division and migration. Monolayer cell cultures, and to a lesser but still substantial extent even three-dimensional organoid systems without vascularization, do not reproduce this architecture. Drug responses measured in the absence of a physiologically realistic stroma are responses to a different physical and chemical environment than the one the drug will encounter in a patient.
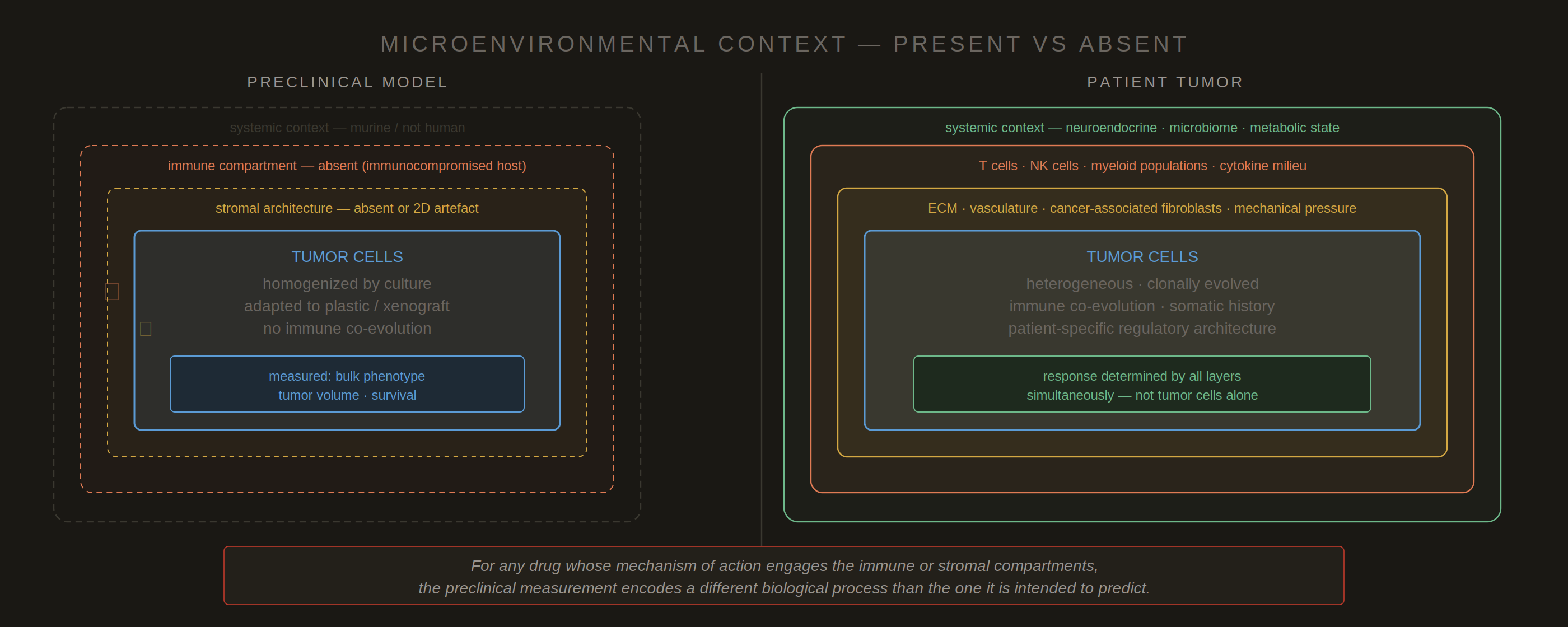
Figure 3. The preclinical model contains only what is technically tractable: tumor cells in a biologically impoverished context. Patient tumor response is governed by all concentric layers simultaneously — immune architecture, stromal organization, vasculature, and systemic metabolic and neuroendocrine state. For any drug whose mechanism of action engages the immune or stromal compartments, the preclinical measurement encodes a different biological process than the one it is intended to predict. This is not measurement noise around a shared signal. It is measurement of the wrong system.
III. What Gets Discarded at the Systemic Level
Pharmacological context and species-specific metabolism
Preclinical models are local. Even the most carefully constructed patient-derived xenograft operates inside a murine metabolic background, with murine cytochrome P450 enzymes, murine plasma protein binding characteristics, murine renal and hepatic clearance mechanisms, and murine systemic inflammatory tone. Drug behavior is not separable from the biological context in which it operates. The compound that produces a given target-site concentration in a mouse may produce a very different concentration in a human, not because the pharmacokinetics were measured incorrectly, but because the measurement was made in a different organism.
Allometric scaling provides a partial correction for some pharmacokinetic parameters. Body weight, surface area, and metabolic rate scale predictably enough across species that exposure predictions have become reasonably reliable for simple molecules with well-characterized metabolism. But allometric scaling cannot correct for qualitative differences in metabolic pathways. A prodrug that requires activation by a human-specific esterase will not be activated in a murine system. A compound primarily cleared by CYP3A4 — which has different substrate selectivity in mice than in humans — will show species-specific exposure profiles that allometric scaling cannot capture because the underlying biochemical mechanism differs.
What gets compressed out at the systemic level is therefore not simply noise around a shared pharmacological signal. It is information about the interaction between the drug’s molecular properties and the specific biological context in which it must operate. That interaction is constitutive of the drug’s clinical behavior. A compound is not pharmacologically characterized by its molecular structure alone; it is characterized by its molecular structure in a biological context, and the preclinical measurement is made in the wrong context.
IV. What Gets Discarded at the Disease Evolution Level
The static measurement of a dynamic process
Cancer in a patient is the product of years or decades of somatic evolution under selection pressure. The genome of a tumor at the time of biopsy is not a random sample from a distribution of possible cancer genomes. It is the survivor of an evolutionary process shaped by the patient’s immune system, the tissue microenvironment, the metabolic demands of progressive growth, and often prior therapeutic interventions. Each of these selection pressures has left traces in the mutational spectrum, the copy number landscape, the epigenetic state, and the transcriptional programs that define the disease.
A cell line or a freshly derived patient-derived xenograft represents a single snapshot of that evolutionary trajectory. What gets compressed out is the trajectory itself: the evolutionary history that explains why the tumor has the vulnerabilities and resistances it has, and the evolutionary plasticity that determines how it will respond to the selection pressure the drug imposes. A drug does not encounter a static molecular target. It encounters a population of cells with the capacity to evolve in response to the selection pressure the drug creates, and the relevant information for predicting clinical outcome is not just the current molecular state but the landscape of accessible evolutionary trajectories from that state.
The target was a snapshot. The disease is a process. The measurement was optimized for the former and is being used to predict the latter.
Cell lines compound this problem substantially. The evolutionary history of a cancer cell line is not the evolutionary history of the patient’s tumor. It is the evolutionary history of the tumor plus the evolutionary history of adaptation to artificial culture conditions: flat plastic surfaces, supraphysiological oxygen tension, defined media with nutrient concentrations that do not resemble interstitial fluid, and the absence of the mechanical and paracrine signals that regulated cell behavior in the original tissue. The selection pressures of cell culture are strong and rapid, and they drive the cell population away from the tumor biology they were meant to represent.
V. What Gets Discarded at the Patient Biology Level
The regulatory architecture that preclinical models cannot represent
The deepest layer of compression, and the most consequential, is also the most difficult to articulate without overstating it. Preclinical models do not compress out specific genes or pathways that could in principle be added back. They compress out the patient-specific regulatory architecture in which those genes and pathways operate, and that architecture is not separable from the disease without destroying the thing you are trying to understand.
A KRAS G12C mutation does not have a fixed clinical meaning. It has a meaning that is conditioned on the genetic background in which it arose, the epigenetic state of the cell that acquired it, the immune evasion programs that co-evolved with it, the metabolic dependencies the tumor developed in the specific tissue microenvironment of this patient’s lung or colon, and the prior selective pressures that shaped the trajectory of this particular clone. A cell line carrying KRAS G12C is not a model of KRAS G12C cancer. It is a model of KRAS G12C cancer in one patient, at one time point, after decades of adaptation to plastic and defined media, in a genetic background that has been shaped by culture selection rather than disease evolution.
The practical consequence is that drug responses measured in preclinical systems are responses to a molecular context that does not exist in any patient — not a simplified version of the patient context, but a different context. The information content of the measurement with respect to the patient-relevant variable is bounded not by measurement noise but by the mutual information between the two biological systems, and for the disease-relevant regulatory programs, that mutual information is often quite low.
This is the argument that cannot be resolved by adding measurement modalities. A multi-omic measurement of a cell line gives you a high-dimensional portrait of a biological system that is not the disease you are trying to treat. More dimensions of the wrong system do not converge on the right answer. The information-theoretic ceiling is not set by measurement bandwidth. It is set by the biological relevance of what is being measured.
Figure 4. Taxonomy of information discarded by standard preclinical measurement, organized by causal distance from clinical outcome. Bar width represents the breadth of discarded information within each category. The five layers are not independent: patient-specific regulatory architecture (Layer V) conditions all layers above it, and microenvironmental context (Layer II) shapes cellular response (Layer I). The deepest layer is also the most completely absent from standard preclinical readouts. The information-theoretic ceiling on clinical prediction from preclinical data is set by the biological relevance of what is being measured, not by the sophistication of the statistical methods applied to it.
The Distortion Metric Was Always There
None of this is to say that preclinical measurement was designed carelessly. The measurement stack that evolved over decades of pharmacological research was optimized — just not for clinical predictive information. It was optimized for reproducibility across laboratories, for regulatory legibility, for experimental tractability, and for the practical constraints of animal husbandry and compound availability. These are real and legitimate considerations. They produced a measurement infrastructure that is extraordinarily good at answering the question it was designed to answer: does this compound produce a detectable and reproducible biological response in a controllable experimental system?
That question is not the same as the clinical question. The distortion metric implicit in the preclinical measurement stack penalizes experimental irreproducibility and rewards mechanistic simplicity. The distortion metric required for clinical prediction penalizes loss of mutual information with patient treatment response, which requires preserving cellular heterogeneity, microenvironmental context, disease evolutionary history, and patient-specific regulatory architecture. These two distortion metrics select for almost orthogonal features of the biological signal.
The data processing inequality closes the argument. Once the clinically relevant information has been discarded, no downstream processing — no machine learning architecture, no foundation model trained on compressed representations — can recover it. The ceiling on predictive performance from preclinical data is not a ceiling set by statistical method. It is a ceiling set by the information content of the measurements themselves, and that ceiling was determined at the moment the measurement protocol was designed.
The implication is not that preclinical models should be abandoned. It is that the measurement design question must be asked explicitly, and answered, before the measurement is made: what distortion metric are we optimizing, and does it correspond to the clinical variable we are trying to predict? Answering that question honestly requires measuring closer to the causal origin of disease, preserving the features that standard compression discards, and accepting that measurements optimized for clinical predictive content will often be noisier, slower, and harder to standardize than the measurements they are meant to replace. That is not a failure of experimental design. It is the correct tradeoff, made visible.
________
On the data processing inequality. For a Markov chain X → Y → Z, the inequality states I(X;Z) ≤ I(X;Y), where I(·;·) denotes mutual information. Applied to the pipeline described here: X is the patient-relevant disease state, Y is the preclinical readout, and Z is any downstream prediction derived from Y. The inequality holds as an equality only when the processing step Y → Z is a sufficient statistic for X, meaning no information relevant to X is discarded. Standard preclinical readouts are very far from sufficient statistics for patient disease state. Rate-distortion theory (Shannon, 1959) formalizes the relationship between compression rate and distortion under a specified distortion function; the optimal codebook depends on the distortion metric in a way that makes compression-metric mismatch a first-order problem, not a second-order correction.